
BEBPA Blog
Volume 1, Issue 3
Mini History of the Potency Bioassay and its Regulatory Drivers
By Laureen Little, PhD, President of BEBPA and Principal Consultant, Quality Services

The concept of potency assays came into being specifically for biopharmaceutical products. Originally, these products were complex and ill-defined mixtures, produced in a natural system. (Like a horse! See photo of Jim below.) The products were not well-understood and rarely purified to any great extent. Sometimes, even the active ingredient was unknown, and our understanding of the Mechanism of Action (MoA) involved much hand-waving. Because of these characteristics these products were defined as a manufacturing process. The mantra “ the product is the process” was a well-known, regulatory concept driving our oversight of biologics for decades.
How did this process = product oversight begin? It started in 1901 with Jim (Figure 1), an ordinary retired milk delivery horse. At that time, the standard treatment for children with diphtheria was an antitoxin serum made from the blood of horses. Jim produced over thirty quarts of antitoxin in three years, but during this time he contracted tetanus. The serum from Jim’s tainted blood was accidentally bottled and used to treat diphtheria patients, causing the death of thirteen children from tetanus. A key problem was the manufacturing in local establishments with no central or uniform controls in place to ensure potency and purity. Nor were there inspections or testing of the final product.
Around the time of the St. Louis deaths, a similar tragedy occurred in Camden, N.J. Nine children died from tetanus after receiving contaminated smallpox vaccine. The news of these incidents flashed across the country within hours, due to the dominance of the cross-country telegraph newly used for communication and news reporting in the US.
Recognizing the critical need for regulatory safeguards, and responding to public pressure, Congress passed the Biologics Control Act in 1902. This act, also known as the “Virus-Toxin Law”, gave the government control over the processes used to make biological products and the responsibility to ensure their safety for the American public.
Vaccines were thus manufactured for several decades, with a focus to assure the safety of the product, but with little knowledge of the potency/efficacy of the batches being released to the public. In the 1940s, Dr. Margaret Pittman in the Biologics Control Laboratory (now CBER) developed the first assay for the pertussis vaccine to assess potency.
This method was an in-vivo mouse assay which included an intracerebral (IC) challenge of the mice with Bordetella pertussis. In this assay, groups of mice are immunized with graded dilutions of vaccine doses and then challenged IC with a lethal dose of B. pertussis. The dose of vaccine at which 50% of the animals are protected is calculated and compared with a reference batch of vaccine. The reference vaccine batch initially used to develop the test was that used in a clinical trial and demonstrated to be efficacious in children.
Dr. Pittman then tested multiple products on the market within 1945-46 and showed that three products had no potency and others varied 10-fold within and between products. The need for assessing potency and not just the safety was obvious from these test results. Manufacturers were required to implement this potency test and by 1949, manufacturers sold whooping cough vaccine approved on potency as well as safety and sterility.
Thus, the modern concept of a functional bioassay, called the potency assay, was born. Most of the early assays were challenge in-vivo assays. Animals (mice, guinea pigs, rats, etc.) are immunized with varying doses of the vaccine and then challenged with the disease organism. Survival rates of immunized animals was compared to animals immunized with a known efficacious batch of vaccine (the reference).
Fast-forward to the 1980s and the biotech industry was born, with the approval of Humulin by the FDA. Eli Lily’s recombinant insulin was made from Genentech’s specially modified bacteria. It was the first drug produced utilizing the modern recombinant DNA technology and among the first genetically engineered products to be available to consumers. Prior to its development, diabetics used insulin isolated from pig and cow pancreases.
Figure 2: Dr. Pittman in the Biologics Control Laboratory. Photo available here.
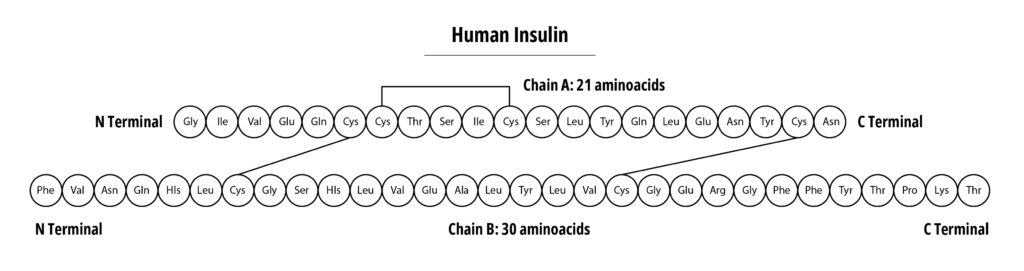
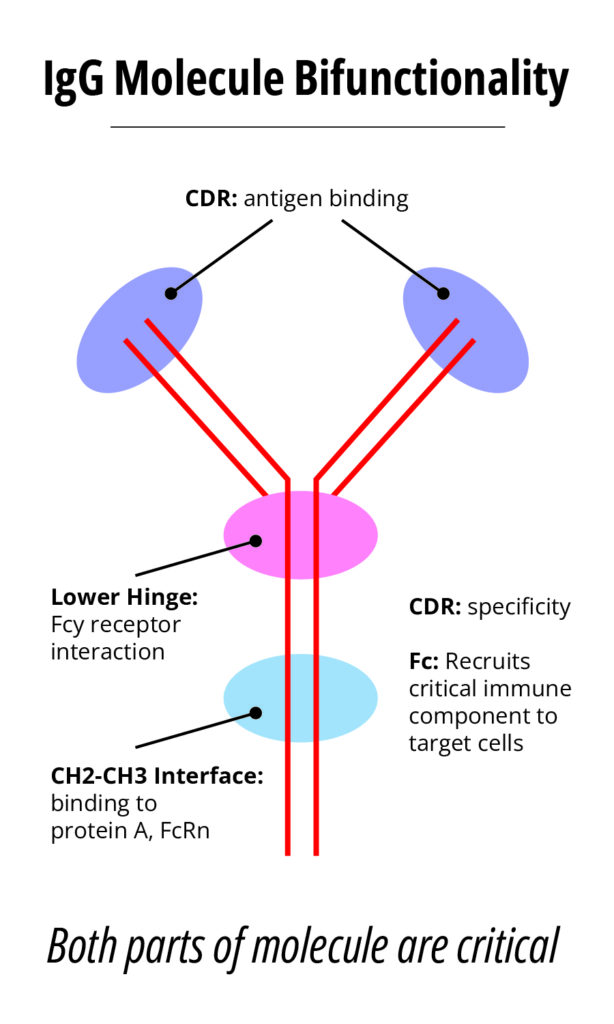
Compare this with the typical monoclonal antibody (mAb) product with a typical Molecular weight of 150 kDa. Insulin can be easily analyzed utilizing a variety of physical/chemical methodologies, this is more difficult for mAbs, especially when determining if the molecule has the appropriate 3-D structure. However, mAbs, have well-known and highly characterized structure function relationships. See figure 4.
The mAb CDR region confers specificity, targeting specific antigens on the cell surface, while the Fc region recruits critical immune component to the cells containing the target antigen. Two common mechanisms of target cell killing accomplished by today’s therapeutic mAb products induce the:
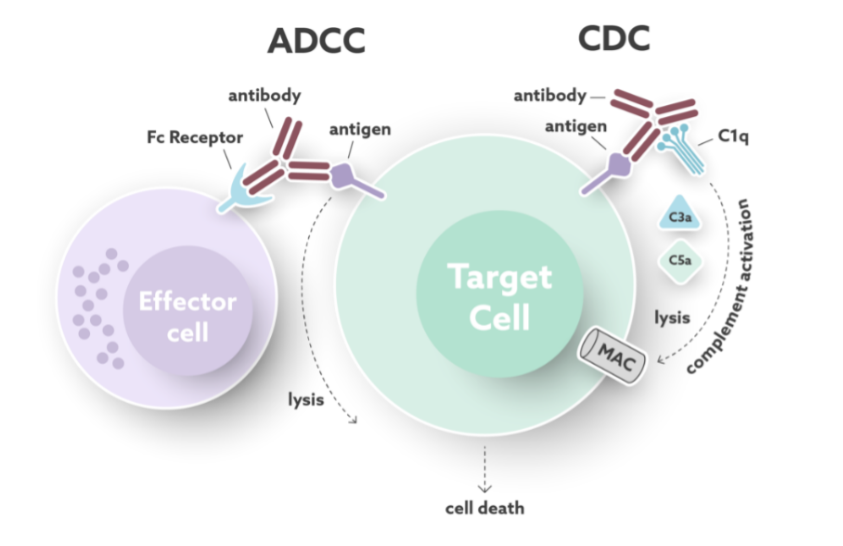
- Antibody Dependent Cellular Cytotoxicity (ADCC) pathway and
- Complement Dependent Cytotoxicity (CDC)
ADCC is induced when the target-bound antibody is recognized by FcγRs on effector cells, such as Natural Killer (NK) cells that cause cell lysis of the target cell. CDC is induced when the target-bound antibody is recognized by C1q from the complement system causing a cascade of events that result in the release of soluble factors and the formation of the membrane attack complex (MAC) that lyses the target cell. See Figure 5.
One of the earliest approved therapeutic mAb products, still on the market, Rituxan, included a CDC potency assay as part of the product release testing. This allowed the assessment of potency for a large molecule with an in-vitro, cell-based bioassay.
Now, we have entered the 2020s and many of our products are again more complex. Instead of a single protein product with a well-defined structure-function relationship, we now have cellular therapeutics with hundreds or thousands of released proteins, some or all of which might be critical to the MoA. The ever-challenging potency assay now becomes increasingly difficult as our products are more complex and the MoAs less defined. A bit like back to the future
These products often contain cells as a drug substance (i.e., active ingredient) that have been isolated, expanded and/or differentiated or even grown to form tissue-like structures in-vitro before use in a patient. Cell-based biotherapeutics can be divided into somatic cell therapy medicinal products and tissue engineered products, depending on their functional principle in the body. Somatic cell therapy medicinal products exert a pharmacological, immunological, or metabolic action to treat, prevent or diagnose a disease, whereas tissue engineered products are intended to regenerate, repair, or replace a human tissue.
As part of the quality control strategy of any cell-based therapeutic, we need to demonstrate that the drug substance is biologically active in the manner needed for a positive clinical outcome. This is where its potency must be evaluated. A potency assay measures the biological activity representing the desired MoA of an ATMP in a quantitative manner. Usually, a product-specific attribute that is directly or indirectly linked to the biological activity is detected. An example of such an attribute is an enzyme whose activity is associated with the relevant biological function of the ATMP such as the ability to interact with existing cells for tissue repair. The challenge when working with such complex systems is to identify an attribute that is quantifiable and represents the relevant MoA of the ATMP.
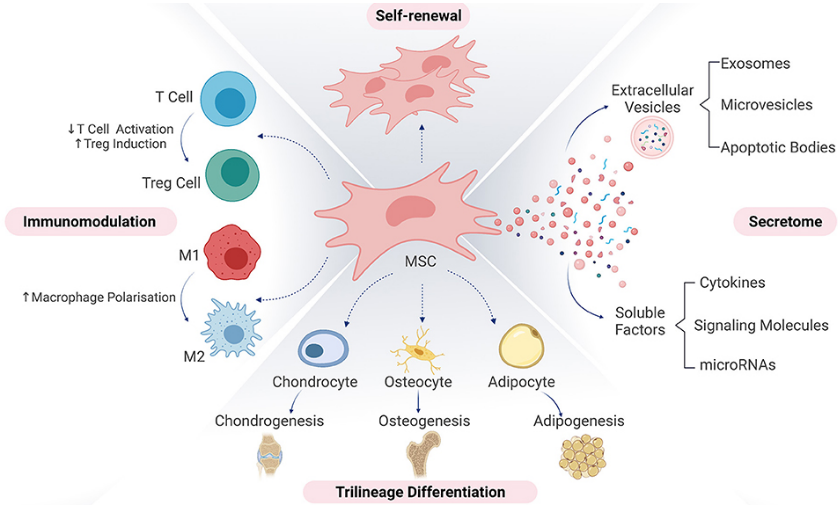
Based upon the potential three MoAs highlighted above, it is reasonable to provide a matrix of potency assays – which cover all the biological pathways. This is not to say that every component/protein must be verified, but that each pathway is biologically active at the expected levels.
Potency assays are a keystone in the manufacture of biotherapeutic products. Whether the products are simple or complex, whether their manufacturing processes utilize old or new technologies, or whether the product is well-characterized or ill-defined, potency is a critical product attribute. To commercialize new products, we must ensure that the routinely manufactured material has equivalent potency as clinical trial material. A critical step in developing the potency bioassay is to understand the disease process we seek to cure. Knowing how our therapeutic product rectifies the body’s physiological responses to this disease and restores the patient to health is necessary to ensure potency. With this knowledge, we can innovate and provide analytical methods which demonstrate that each released product batch has the appropriate biological activity to be efficacious.

About The Author: Laureen Little, PhD
Dr. Laureen Little has over 30 years of biotechnology experience. She is the president of Biopharmaceutical Emerging Best Practices Association (BEBPA; www.bebpa.org) a non-profit organization she founded in in 2007 to promote scientific conferences for scientists working in the biopharmaceutical industry. BEBPA now hosts 3 to 4 conferences annually on such topics as potency bioassays, Host Cell Proteins, Immunogenicity assays, automation in analytical laboratories and the handling of reference materials. The organization has grown over the past decade and is now recognized as the venue for frank, open discussion and has resulted in vast improvements in technical approaches in various aspects of analytical development.
She is also a principal consultant with Quality Services, specializing in biological assay optimization and validation. She has been an instructor for FasTrain Courses (www.traincourses.com), for more than 20 years teaching various GXP courses as well as a popular potency assay course.
Laureen has worked with many firms developing, qualifying, and validating biological potency assays for BLA submissions, Pre-Approval Supplements for commercial products and special amendments to support facility/manufacturing changes. The various products she has worked with include: monoclonal antibodies, rDNA products, peptide hormones, CBER regulated devices, gene therapy products, autologous cell therapies, toxins, cancer vaccines, viral vaccines, prophylactic vaccines, blood products, enzyme therapeutics, anti-angiogenic drugs, and lipid-based therapies. The potency assays have included animal model systems, cell-based methods, and binding assays.