
BEBPA Blog
Volume 2, Issue 3
Beyond Potency: Leveraging Bioassays for Contaminant Testing
By Jamison Grailer, Sr. Research Scientist, Promega Corporation
Bioassays are used extensively for potency determination of biologic therapeutics. However, ensuring the purity and safety of biologic drugs is of similarly vital importance. Biologic drugs are highly complex and impurities can arise during production, purification, and storage. Unlike small-molecule drugs, biologics are susceptible to a wide range of contaminants—including host cell proteins, endotoxins, and other bioactive substances that can affect their safety, efficacy, and stability. Detecting these impurities requires robust analytical tools, and bioassays can play a central role in the quality control of these products.
Scientists have leveraged biological systems for the sensitive detection of contaminants that are often challenging to quantify through traditional means. Among the most widely utilized examples is the Limulus Amebocyte Lysate (LAL) assay for detecting lipopolysaccharides (LPS), a potent endotoxin commonly found in gram-negative bacterial cell walls. Endotoxin contamination of injectables and medical devices can trigger severe inflammatory responses, fever, and in extreme cases, septic shock and death. The LAL assay utilizes an extract from horseshoe crab blood that reacts specifically with LPS. In horseshoe crabs, this reaction causes coagulation, effectively walling off the infection. In the LAL assay, the coagulation generates a quantitative signal that reflects the presence of LPS in the test sample. Due to its specificity and sensitivity, the LAL assay became the industry standard for endotoxin testing in nearly all biologic products, illustrating the power of adapting naturally evolved defense mechanisms for analytical purposes.
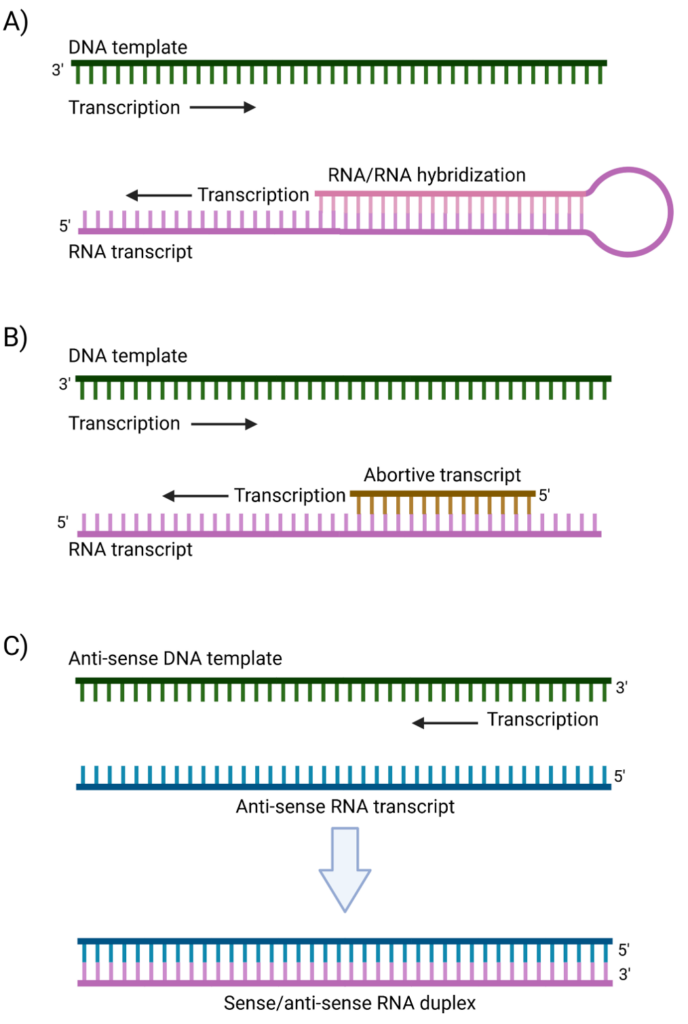
The emergence of mRNA as a therapeutic modality has led to new challenges in contaminant testing. Large-scale production of single-stranded RNA (ssRNA) for mRNA-based therapeutics and vaccines is primarily achieved through in vitro transcription (IVT), an enzymatic process designed to generate high-yield, functional mRNA transcripts that serve as the drug substance. However, like any biological process, IVT is inherently imperfect and produces unintended byproducts, including double-stranded RNA (dsRNA). As the name suggests, dsRNA is composed of complementary RNA stretches that anneal to form duplexed structures. The formation of dsRNA is largely driven by T7 RNA polymerase (T7 RNAP), the enzyme most commonly used in IVT, through several distinct mechanisms. One major pathway involves the intrinsic RNA-dependent polymerase activity of T7 RNAP, where the 3′-end of a newly synthesized transcript hybridizes with complementary sequences on the same or another RNA molecule, serving as a primer for further elongation and leading to dsRNA formation (Ref 1, Figure 1A). Additionally, T7 RNAP is prone to transient stalling, leading to the generation of truncated abortive transcripts that can anneal to complementary sequences and initiate unintended RNA-dependent extension (Ref 2, Figure 1B). Another key mechanism is template switching or “jumping,” wherein the polymerase disengages from its template and rebinds to an anti-sense strand or another segment of the template, generating complementary RNA products that subsequently anneal into dsRNA structures (Ref 3, Figure 1C).
These dsRNA contaminants pose a significant challenge for IVT-derived therapeutics, as they are recognized by several key cellular pattern recognition receptors (PRRs), including Toll-like receptor 3 (TLR3), retinoic acid-inducible gene I (RIG-I), and melanoma differentiation-associated protein 5 (MDA5) (reviewed in 4). TLR3, primarily located within endosomes, detects endosomal dsRNA, whereas RIG-I and MDA5 operate in the cytoplasm. Upon binding dsRNA, these receptors launch signaling cascades, culminating in the activation of transcription factors such as NF-κB, interferon regulatory factor 3 (IRF3), and IRF7. The net effect is upregulation of pro-inflammatory cytokines and type I interferons, creating a potent antiviral state that promotes the activation of RNA-degrading enzymes and triggers immune effector functions aimed at eliminating the perceived viral infection. When this pathway is inadvertently activated by dsRNA impurities in therapeutic mRNA preparations, the response can destabilize the delivered mRNA and diminish its translation, partially through enzymatic degradation and the inhibition of translation machinery. This not only lowers transgene expression but can also provoke unwanted immunogenicity, ultimately reducing both the safety and efficacy of mRNA-based therapeutics. Understanding and mitigating dsRNA formation is therefore critical to improving the safety and efficacy of IVT-derived mRNA therapeutics.
Key strategies to minimize dsRNA have arisen to both reduce the formation of dsRNA during IVT as well as the removal of dsRNA after IVT. They begin with careful optimization of the IVT reaction parameters, including the fine-tuning of magnesium, salt, and nucleotide concentrations to reduce polymerase stalling and off-target elongation (5). Additionally, using high-fidelity or engineered T7 polymerase variants can limit dsRNA (6). Rational sequence design of the transcript (e.g., avoiding complimentary motifs and eliminating sequence that may result in secondary structure) further reduces the risk of dsRNA formation. Post-IVT, dsRNA removal strategies are typically grounded in chromatographic techniques such as ion-exchange and reverse-phase HPLC. Solid-phase extraction using cellulose or specialized dsRNA-binding resins provides an additional platform for removing dsRNA, exploiting the different binding affinities of ssRNA versus dsRNA under defined salt and pH conditions. The goal of these combined approaches is to enrich the final product for ssRNA, significantly reducing dsRNA impurities and enhancing the safety and efficacy of mRNA-based therapeutics.
Even with modern mitigation strategies and purification steps, residual dsRNA can persist at low levels in mRNA therapeutics, making accurate quantification critical for product safety and regulatory compliance. Currently, enzyme-linked immunosorbent assays (ELISAs) and dot blots using dsRNA-specific monoclonal antibodies (such as J2 or K1) are common approaches because they are relatively straightforward, accepted technologies. However, these antibody-based methods can suffer from several limitations. First, their performance depends heavily on the antibodies’ specificity and affinity, which may vary with different RNA sequences or structures. Furthermore, these antibodies can display sequence-dependent binding, raising the risk of underestimating dsRNA (7). Finally, these methods often require time-consuming protocols and complex sample preparation. These challenges highlight the need for an antibody-independent and RNA sequence-independent detection method that offers accurate, precise, and sensitive dsRNA quantification.
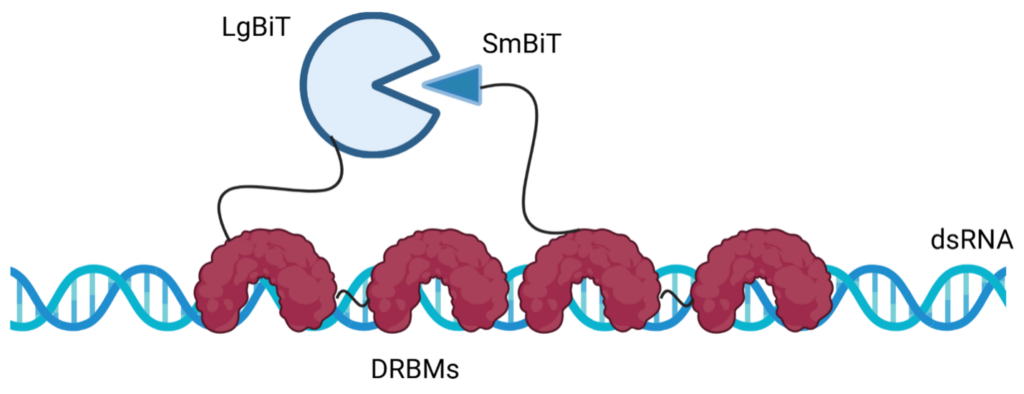
At the 2024 European BEBPA conference, Promega introduced an innovative, antibody-independent method of dsRNA detection and quantitation. Similar to the LAL method, we leveraged naturally occurring anti-viral defense mechanisms to detect dsRNA. The new assay combines evolutionarily-adapted dsRNA binding proteins with the sensitivity of NanoBiT® (NanoLuc® Binary Technology). The NanoBiT® system is a split-luciferase reporter technology consisting of two subunits, Large BiT and Small BiT, which reconstitute functional luciferase when brought into proximity, generating a quantifiable luminescent signal (8). The high affinity and specificity of naturally-occurring dsRNA binding motifs (dsRBMs) make them ideal for detecting dsRNA in this assay. By fusing dsRBMs to the NanoBiT® luciferase subunits, the assay detects dsRNA through the reconstitution of luciferase activity when dsRNA binding occurs, producing a luminescent signal that directly correlates with dsRNA concentration (Figure 2). This system allows for precise, luminescence-based quantification of dsRNA in IVT drug substances. The assay is sensitive and specific, user-friendly, and adaptable to GMP standards; providing a robust tool for ensuring the quality and safety of mRNA therapeutics by enabling reliable detection and quantification of residual dsRNA in the production workflow.
While bioassays are most often associated with drug potency testing, their application in impurity detection may offer unique advantages, potentially capturing the nuanced biological impacts of contaminants that conventional technologies may miss. By leveraging the unique capabilities of biological systems—whether through cell-based assays, or evolutionary defense mechanisms such as those used in the LAL and the NanoBiT® dsRNA Detection Assay—these tools provide innovative approaches to detect and quantify impurities at levels that safeguard patient health. As biologic therapies become increasingly complex, it is essential to ensure that this innovation in therapeutic design is matched by equally rigorous innovation in bioassay technology, both for potency testing as well as other critical measures of product safety and efficacy.
For more information, please visit:
References:
- Arnaud-Barbe, N., V. Cheynet-Sauvion, G. Oriol, B. Mandrand, & F. Mallet. (1998) Transcription of RNA templates by T7 RNA polymerase. Nucleic Acids Res. 26: 3550-3554.
- Gholamalipour, Y., A. Mudiyanselage, & C. Martin. (2018) 3’ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character – RNA-Seq analyses. Nucleic Acids Res. 46: 9253-9263.
- Mu, X., E. Greenwald, S. Ahmad, & S. Hur. (2018) An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 46: 5239-5249.
- Chen, Y., & S. Hur. (2022) Cellular origins of dsRNA, their recognition and consequences. Nat Rev Mol Cell Biol. 23: 286-301.
- Guo, L., Z. Liu, S. Song, W. Yao, M. Yang, & G. Chen. (2024) Maximizing the mRNA productivity for in vitro transcription by optimization of fed-batch strategy. Biochem Engineering Journal. 210: 109412.
- Dousis, A., K. Ravichandran, E. Hobert, M. Moore, & A. Rabideau. (2023) An engineered T7 RNA polymerase that produces mRNA free of immunostimulatory byproducts. Nat Biotechnol. 41: 560-568.
- Bonin, M., J. Oberstrass, N. Lukacs, K. Ewert, E. Oesterschulze, R. Kassing, & W. Nellen. (2000) Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. RNA. 6: 563-570.
- Dixon, A., M. Schwinn, M. Hall, K. Zimmerman, P. Otto, T. Lubben, B. Butler, B. Binkowski, T. Machleidt, T. Kirkland, M. Wood, C. Eggers, L. Encell, K. Wood. (2016) NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem Biol. 11: 400-408.

About The Author: Jamison Grailer
Jamison Grailer is a Senior Research Scientist at Promega Corporation, where he leads the R&D Bioassay Development Team. His team specializes in harnessing Promega’s cutting-edge technology to create innovative bioassay solutions that drive the discovery and development of biological therapeutics. Dr. Grailer has authored over 40 peer-reviewed publications and contributed to four book chapters, with his research spanning immunology, oncology, materials science, and biotechnology. Prior to joining Promega, he completed postdoctoral training in innate immunity and inflammation at the University of Michigan Medical School.