
BEBPA Blog
Considerations for manufacturing ex-vivo therapeutic products: CRISPR, Cell Sourcing, Potency and Beyond
By Laureen Little, PhD, President of BEBPA and Principle Consultant Quality Services
The first therapy using the CRISPR technology was approved in the UK on 21 November 2023, Casgevy (exagamglogene autotemcel) now has a conditional authorization from the UK Medicines and Healthcare products Agency (MHRA). This authorization is for two indications, Sickle Cell Disease (SCD) and transfusion-dependent β-thalassemia (TDT). The sponsors for Casgevy were Vertex Pharmaceuticals and CRISPR Therapeutics Casgevy is also under review by the European Medicines Agency (EMA), the Saudi Food and Drug Authority, and the U.S. Food and Drug Administration (FDA). The FDA granted Priority Review for SCD and Standard Review for TDT and assigned Prescription Drug User Fee Act (PDUFA) target action dates of December 8, 2023, and March 30, 2024, respectively. The clinical trial for the treatment of SCD included 29 trial participants of which 28 reached their primary outcome of becoming free from severe vaso-occlusive crises.
Molecular Basis of Sickle Cell Disease (SCD)
SCD is an inherited blood disorder that affects red blood cells (RBC). In SCD, the oxygen-carrying protein in red blood cells, hemoglobin, is comprised of two α-chains and two β-chains. SCD patients carry a point mutation in the β-globin gene, which leads to a glutamate-to-valine substitution at position 6. This change results in a structure that forms rigid polymers when oxygen is not bound. This causes RBCs to adopt a characteristic sickle or crescent shape, thus the name of the disease. The altered RBC shape also predisposes them to adhere to capillary walls causing blockages in blood vessels. These blockages result in a lack of oxygen in nearby tissues and painful vasculo-occlusive crises (VOC) in patients. The primary endpoint in the Casgey clinical trial was the elimination of these VOCs.
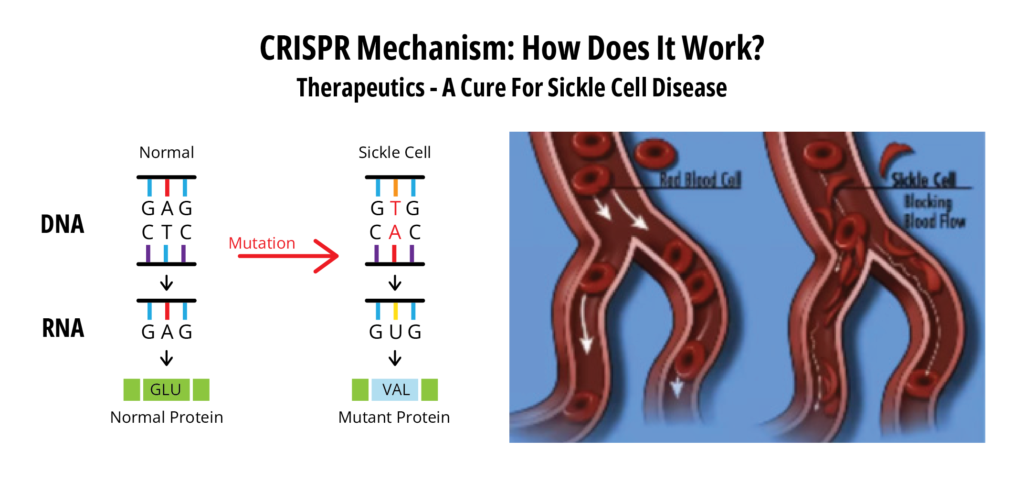
(Reference 1)
Interestingly, Casgevy does not fix the above point mutation in sickle cell RBCs. Instead, it compensates for the loss of adult hemoglobin by inducing fetal hemoglobin, the main oxygen carrier in the fetus (Reference 2). Expression of the fetal hemoglobin is normally switched off shortly after birth via transcription repressors (BCL11A) of the γ-globin gene. The CRISPR components of Casgevy interfere with the transcription repressors and result in the resumption of fetal hemoglobin expression in the treated CD34+ cells.
Casgevy is, therefore, an autologous ex-vivo cellular therapeutic product. The manufacturing process starts with the isolation of CD34 + hematopoietic stem cells from patient bone marrow. CRISPR editing components are then introduced in the lab by electroporation as a ribonucleoprotein complex, comprised of a synthetic guide RNA and a Streptococcus pyogenes Cas9 endonuclease. The edited cells are then infused back into the patient, allowing the body to produce functioning fetal hemoglobin.
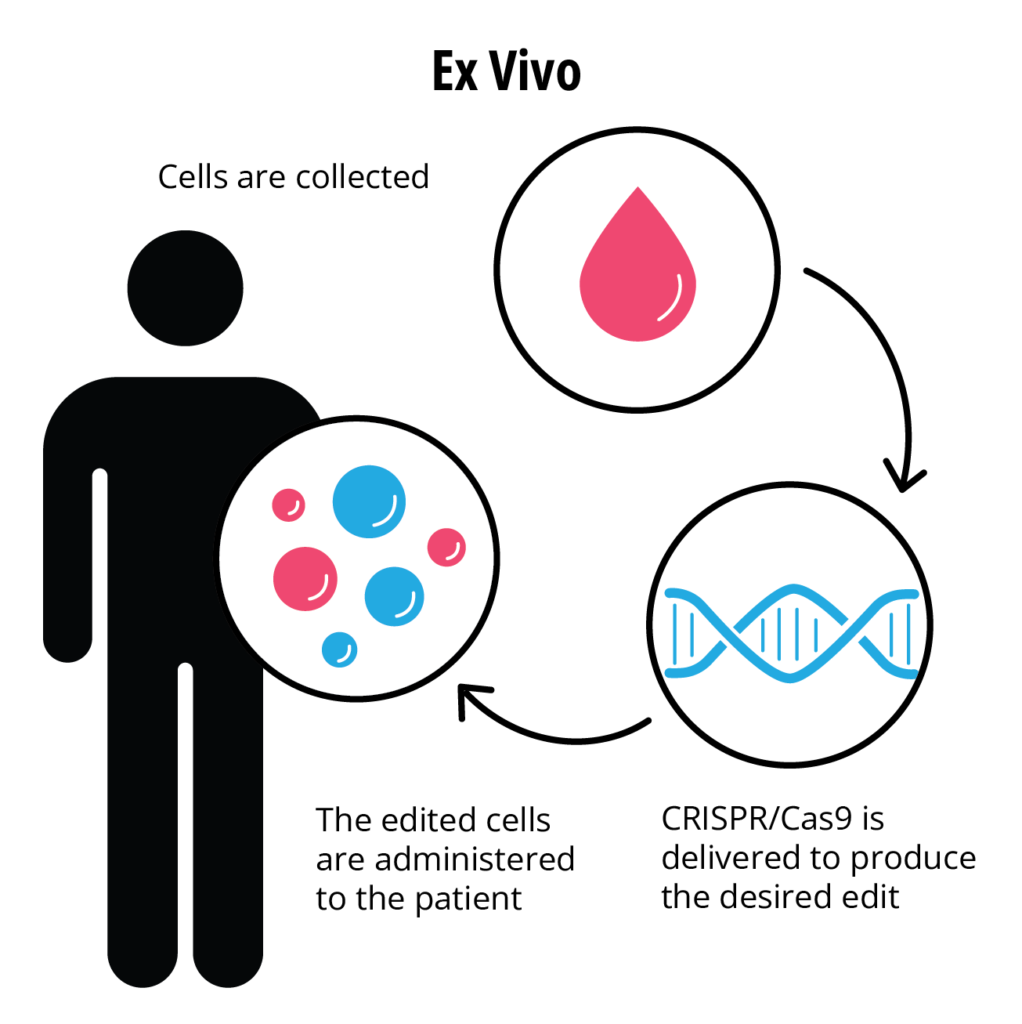
(Reference 3)
Casgevy, at its simplest, is the newest ex-vivo gene therapy using autologous CD34+ cells. Prior approved ex-vivo products utilize retroviruses for inserting working genes into patient cells rather than editing the existing cellular DNA. These retrovirus gene therapies include:
- Strimveli, treatment for severe combined immunodeficiency due to adenosine deaminase deficiency (ADA-SCID (Reference 4) Approved 2016
- Zynteglo, treatment for beta thalassaemia (Reference 5) Approved 2019
- Libmeldy, a treatment for metachromatic leukodystrophy (MLD) (Reference 6) Approved 2020.
For these ex-vivo gene therapy products the potency testing usually includes cell viability and expression of the transgene protein, but also % vector positive cells, vector copy number and transduction efficiency. However, functional potency of transduced CD34+ cells may be difficult to address in vitro after manufacturing, as part of the biological activity takes place only after administration into the patient. CD34+ cells are hematopoietic stem cells, which are expected to find their way back to the bone marrow and engraft. Therefore, an in-vitro potency assay will need to mimic the critical steps of this engraftment, proliferation, and differentiation.
Potency assays are considered a critical CMC requirement for biotherapeutics, and it is expected that ex-vivo cellular therapeutics will be no exception. A couple of years ago FDA placed a hold on another autologous CD34+ hematopoietic stem cell product edited at the HBG1 and HBG2 gamma globin promoter genes because of the need for an improved potency assay.
Measuring the functional potency of transduced CD34+ cells in clinical samples post treatment such as using bioanalytical tools to measure engraftment capacity (e.g., colony formation assay), differentiation into various cell lineages (FACS) and expression level/quality of the transgene product is clearly not a release assay. These assays are instead analytical biomarkers for product efficacy after treatment.
Five-minute tutorial on CRISPR technology
For those of us not familiar with the CRISPR technology there are several short videos available on the internet (References 1 and 7). These videos provide entertaining and informational tutorials on the technology. CRISPR is made up of two key parts: a CRISPR-associated (Cas) nuclease, which binds and cuts DNA, and a guide RNA sequence (gRNA), which directs the Cas nuclease to its target.
These two components were discovered in bacterial immune systems, where they protect the bacteria from invading viruses by cutting the viral DNA and disabling them. Once the molecular mechanism for its DNA-cleaving ability was discovered, it was quickly developed as a tool for editing genomes.
CRISPR technology was developed by Dr. Jennifer Doudna and Dr. Emmanuel Charpentier. Their groundbreaking paper, revealing that the CRISPR-Cas9 bacterial immune system could be repurposed as a gene editing tool, was published in the journal Science in 2012. (Refence 8) It wasn’t until 2020, well after the technology had been adopted in labs around the world, that Doudna and Charpentier won the Nobel prize in Chemistry for their discovery, becoming the first all-female team to do so.
Definitions for CRISPR include:
- CRISPR: “Clustered Regularly Interspaced Short Palindromic Repeats” of genetic information that some bacteria use as part of an antiviral system and that Dr. Charpentier and others discovered how to use as a gene-editing tool
- Cas9: a CRISPR-associated (Cas) endonuclease, or enzyme, which acts as “molecular scissors” to cut DNA at a location specified by a guide RNA
- Guide RNA (gRNA): a type of ribonucleic acid (RNA) molecule that binds to Cas9 and specifies, based on the sequence of the gRNA, the location at which Cas9 will cut DNA

(Reference 8)
Unique CMC Issues of ex-vivo cellular therapeutics
One unique difference of any ex-vivo cellular therapeutic, is the sourcing of donor human cells as a starting material. Questions are being raised about how to appropriately qualify these cells. It is suggested that manufacturers focus on the following:
- Safety testing
- Sterility and mycoplasma testing is recommended
- Allogenic cells require screening for additional relevant human pathogens not included in donor eligibility testing
- Establish acceptance criteria for incoming material
- Minimum cell number
- %CD cells types as required for the product (CD34+, CD3, etc.)
- Viability
- Conduct additional characterization studies
- Phenotypic analysis (for example % and absolute number of CD4+ and CD8+, NK monocytes, B cells, and so on )
- Note that the above analyses may inform process development on the need for enhanced cell selection
From a regulatory perspective the starting cells are typically sourced from either a medical clinic or a donor tissue facility. It is critical for sponsor companies to realize that these are not GMP facilities. The latter, are usually regulated in the US by 21CFR Parts 1270 and 1271 (AKA the Good Tissue Practices). It is the responsibility of the therapeutic manufacturer to confirm the cell sourcing facility maintains good sterile practices designed to reduce potential contamination with adventitious agents and provides traceability and appropriate documentation. Once the cells are received by the manufacturer, they are now regulated by the appropriate cGMP practices (usually the drug regulations 21CFR Parts 210/211 or the biologic regulations in Part 600 or the device regulations in Part 820).
Safety Issues with ex-vivo cellular therapies
One of the biggest questions about all genetic therapeutics is the potential for long term safety issues arising from the introduction of genetically altered cells into the body. Many of these cells become a permanent addition to the patient physiology. The FDA recently announced (11/28/2023) that they received reports of T-cell
malignancies, including chimeric antigen receptor (CAR)-positive lymphoma, in patients who received treatment with B-Cell maturation antigen (BCMA) or CD19-directed autologous CAR T cell immunotherapies. (Reference 9) FDA determined that the risk of T-cell malignancies is applicable to all currently approved immunotherapies of this type. T-cell malignancies occurred in patients treated with several products. Currently approved products in this
class (listed alphabetically by trade name) include:
- Abecma (idecabtagene vicleucel)
- Breyanzi (lisocabtagene maraleucel)
- Carvykti (ciltacabtagene autoleucel)
- Kymriah (tisagenlecleucel)
- Tecartus (brexucabtagene autoleucel)
- Yescarta (axicabtagene ciloleucel)
This finding will undoubtedly increase the focus and need for following patients receiving any form of genetically modified cells in long term safety studies. Current approved products have been required to conduct 15-year, long term follow-up observational safety studies, to assess the long-term safety and the risk of secondary malignancies occurring after treatment. It is also expected that patients will be monitored life-long for new malignancies by their oncology teams as part of their on-going healthcare practices. As yet, there is no discussion about pulling these products off the market, but we expect to see more risk/benefit analyses being made public as the numbers continue to roll in.
- Illustration from video available at https://www.synthego.com/learn/crispr#:~:text=The%20CRISPR%20system%20is%20the,DNA%20known% 20as%20a%20protospacer.) in an interview with Jennifer Doudna, PhD., one of the inventors of the CRISPR technology and co-winner of the 2020 Nobel prize (Professor at UC Berkeley)
- https://mhraproducts4853.blob.core.windows.net/docs/899345634964c32033efbf909906284c5859a9cd)
- Potency testing of cell and gene therapy products, P. Salmikangas, B Carlsson, C Klumb, T Reimer, S Thirstrup Regulatory Science Volume 10 – 2023 May https://doi.org/10.3389/fmed.2023.1190016
- European Public Assessment Report for Strimvelis, (2016). Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/strimvelis)
- European Public Assessment Report for Zynteglo, (2019). Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/zynteglo)
- European Public Assessment Report for Libmeldy, (2020). Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/libmeldy),
- https://crisprtx.com/#
- Martin Jinek et al., A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337, 816-821(2012). DOI: 10.1126/science. 1225829)
- https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/fda-investigating-serious-risk- t-cell-malignancy-following-bcma-directed-or-cd19-directed-autologous)