
BEBPA Blog
Product Potency Standards Throughout The Life Cycle of the Potency Assay
The determination of the potency of the RS throughout a product’s lifecycle needs to be accurate, precise, reliable, and consistent across time and across multiple batches of reference standard, even in the face of changing characteristics of the dose-response curve. This is a particular challenge early in product development when often only Interim Reference Standards (IRS) are available and no higher-order standards (e.g., a Primary RS) are available for comparison throughout the development cycle.
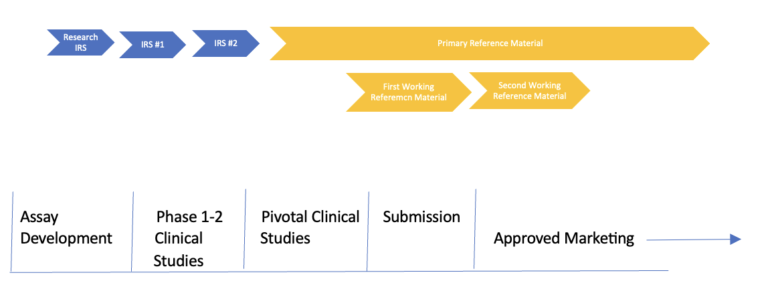
Most potency assays evolve during product development, which adds another layer of complexity. Thus, we have changing product characteristics during development and simultaneously the addition/subtraction/improvement of the potency assay. We often start with a single non-GMP batch of product material, inherited from a research group, which is utilized during early assay development. This material is often used to develop our early potency assays, but not release clinical material. As the manufacturing process is developed and early phase clinical trial material (CTM) is manufactured, we often find ourselves needing a second IRS. This might be due to a lack of volume or changes in the biological dose-response curve of the CTM compared to the research material.
As we move through the PI/II/III manufacturing changes, it is always important to compare and contrast the various lots of IRS used to release CTM. If feasible, it would be simplest to utilize the same IRS for all the CTM batches. However, as mentioned above, this is rarely possible, due to either volume restrictions or changes in the dose-response curves. When one must change from one IRS to another IRS it is critical to provide head-to-head comparisons to bridge the various IRS. This is critically important to ensure that all the clinical batches can be utilized to determine the appropriate potency specification prior to commercialization.
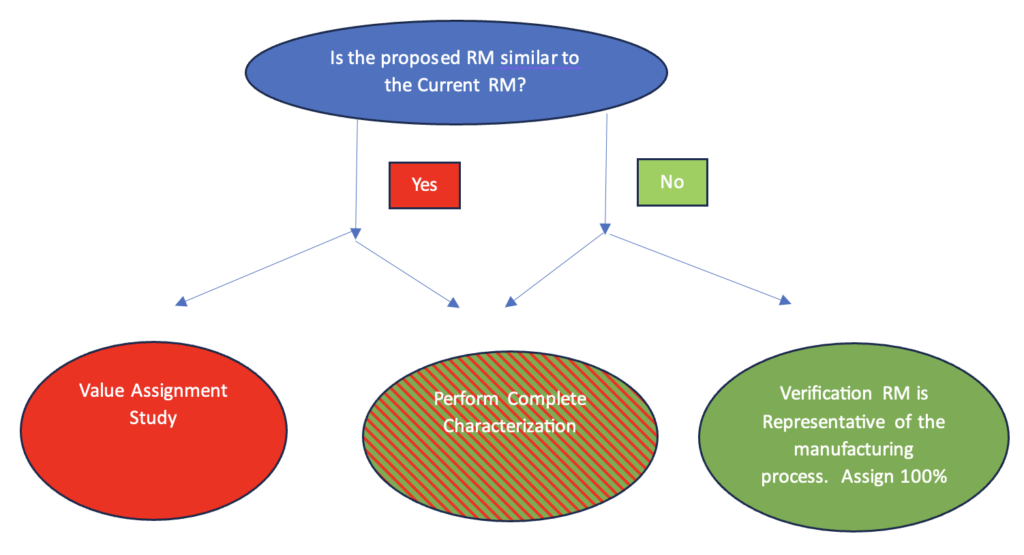
If two interim reference material batches, say the first non-GMP batch and the proposed reference material to be used to release clinical trials are similar, if the dose-response curves are in fact similar, the bridging study is straightforward. (See Figure 2) The bridging is usually a formal study described in a written protocol. The protocol typically includes both product characterization and statistical studies for value assignment of the new IRS. There are many approaches for determining this value assignment protocol. It will involve understanding the precision, bias, and accuracy of the current bioassay used to bridge the two IRS batches.
The most common question asked by bioassayists at this juncture is “How many runs do we need to do?” Unfortunately, there is no set and established answer to this question. The answer depends greatly on the assay precision and bias as suggested above as well as the clinical requirement for equivalence. The best advice is to work with a statistician to determine the necessary power of the bridging experiments. As a first step ask yourself and your clinical colleagues how “wrong” can I be in the potency value assignment and not impact the clinical trial? If I assign a value which might be 10% high or low – does this result in releasing material which is no longer to be efficacious? Or is it so potent it could cause patient safety issues? If you answer yes to this question, then you know that ± 10% is too wide of a confidence interval. Continue asking yourself this question until you find an acceptable range. Give this information to your statistician along with the precision and bias characteristics of the method and they can calculate the required number of runs.
If a manufacturing change results in a change of the dose-response curve of the proposed RS it will be impossible to value assign the new reference from the old reference. (See Figure 2) In this case it is critical to demonstrate that the old and proposed RS dose-response curves are in fact different. This is typically done by running the two lots in a head-to-head study. A head-to-head study is strongly recommended, as during this time frame the potency assay is often evolving and the dose-response curves might change between assay versions. Next carefully determine that the proposed RS is representative of material to be manufactured from this point forward. Once these two items are demonstrated satisfactorily, a full characterization of the new RS should be carried out and a comparison to the prior reference material lot made. Once this is accomplished, the new batch is usually given a value of 100%. All new RS lots will now be bridged to this batch rather than the earlier RS with different dose-response curve characteristics.
It is critical that all IRS used to release CTM are bridged. This is data almost always required by regulators. It is best to involve product experts, bioassay experts and statisticians in the design of these bridging exercises.
Several key RS activities happen in late clinical trials and just prior to submitting an application/dossier for approval. Firms need to draft final protocols for supporting the maintenance of their reference material (RFM) post commercialization. Although there is some regulatory guidance for maintaining reference material, many aspects of a good reference program are driven by good science and a thorough understanding of the patient and manufacturing risk if one does not have a rigorous RFM program in place. Typical requirements for the RFM program include the following elements:
- Establishment of a two-tier reference program.
- A two-tiered system consists of a primary reference standard (PRS), sometimes called a gold standard and a working reference standard (WRS) sometimes called a secondary standard.
- The PRS is used to qualify all future WRS. It is not used for routine product release. It is best if a product batch which is known to be efficacious is laid down as the PRS.
- The WRS typically comes from the standard manufacturing stream and is qualified against the PRS. It is used on a daily basis to release product. Sometimes the very first WRS is from the same product batch as the PRS.
- During the development process described above, the various IRS batches are value assigned using a daisy chain, that is they are value assigning using the current IRS being used to release clinical material. This approach changes at the end of the pivotal trial. Now all future Working Reference Standard (WRS) are qualified, and value assigned using this PRS. See Figure 3.
- WRS qualification protocol.
- This includes a description of how to select, completely characterize, and value assign the potency of the candidate WRS from the PRS.
- Reference Stability Program.
- The PRS and WRS must both have ongoing real-time stability programs. The testing timepoints and details of what is to be reported is outlined in detail in the testing protocols.
- Reference Monitoring Program
- Both the PRS and WRS are usually monitored. Formal trending data is typically collected and assessed semi-annually or annually. QC samples, dose-response curves and the potency of recently manufactured lots are often part of this annual trending.

At recent BEBPA Bioassay Conferences, we polled the audience to find out current practices for laying down and establishing product reference material for bioassays. We found that most of us do not have available international references, and therefore use either an engineering lot or a pivotal clinical batch for our PRS. This is not surprising given the innovative nature of our current product pipeline.
More interesting is the result from our more recent polls indicating progress toward implementation of two-tiered reference programs. In 2019, we started asking bioassay conference attendees if they use a two-tier reference system and found that 50% of the audience had only a working reference material. We ask the question annually and have seen a consistent increase in the number of two-tier programs. In 2023 only 30% stated they only had one-tier systems.

In conclusion, during the product life cycle, it is always critical to pay close attention to any potency RS which is used to release product for use in the patient population. If releasing CTM, it is critical to bridge all the IRS and understand the evolution of the batches and whether they are or should be equivalent. If a proposed IRS is not equivalent to a current IRS a clear picture should be established as to what the changes are, and if possible, a clear explanation as to why they are behaving differently. Additionally there should be a clear demonstration that the new IRS is representative of expected future CTM batches. During commercialization the product should no longer be changing as the process is now defined, in this case a PRS should be manufactured and used to ensure that future WRS and PRS are equivalent to the first PRS and therefore equivalent to the material used to demonstrate product efficacy. Close attention to these aspects of the RS will ultimately pay off in assurance of product safety and efficacy as well as save valuable time and resources during development.
- ICH Q6B: Specifications: Test Procedures and Acceptance Criteria for Biotechnological/ Biological Products. March 1999. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q6B/Step4/Q6B_Guideline.pdf
- FDA Guidance for Industry: Q7 Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients. September 2016. Revision 1. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073497.pdf
- Official USP Reference Standards
https://www.usp.org/reference-standards
- FDA Guidance for Industry: INDs for Phase 2 and Phase 3 Studies Chemistry, Manufacturing, and Controls Information. May 2003
https://www.fda.gov/downloads/Drugs/Guidances/ucm070567.pdf
- European Pharmacopoeia 7.0 – Reference Standards. 01/2008
http://www.fptl.ru/biblioteka/standartnye-obrazcy/EP-7_Reference-standards.pdf
- Recommendations and Best Practices for Reference Standards and Reagents Used in Bioanalytical Method Validation. Joseph F. Bower, Jennifer B. McClung, Carl Watson, Takahiko Osumi, and Kátia Pastre. The AAPS Journal 2014 Mar, 16(2): 352-356
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3933579/
- Reference Standards for Therapeutic Proteins: Current Regulatory and Scientific Best Practices (Part 1). Mire-Sluis A, Ritter N., Cherney, B., Schmalzing, D., and Blumel M. BioProcess Intl., March 1, 2014
- Reference Standards for Therapeutic Proteins (Part 2). Mire-Sluis, A. BioProcess Intl. May 1, 2014 https://bioprocessintl.com/2014/reference-standards-for-therapeutic-proteins-351584/
- Potency assignment of biotherapeutic reference standards Faya P, Borer MW, Griffiths KL, Parekh BS.. J Pharm Biomed Anal. 2020 Nov 30;191:113577. doi: 10.1016/j.jpba.2020.113577. Epub 2020 Aug 24. PMID: 32891042.
- The First WHO International Standard for Harmonizing the Biological Activity of Bevacizumab. Jia H, Harikumar P, Atkinson E, Rigsby P, Wadhwa M. Biomolecules. 2021 Oct 30;11(11):1610. doi: 10.3390/biom11111610. PMID: 34827607; PMCID: PMC8615914.
- International standards for monoclonal antibodies to support pre- and post-marketing product consistency: Evaluation of a candidate international standard for the bioactivities of rituximab. Prior S, Hufton SE, Fox B, Dougall T, Rigsby P, Bristow A; participants of the study. MAbs. 2018 Jan;10(1):129-142.
https://www.ncbi.nlm.nih.gov/pubmed/28985159 - A comparison of vials with ampoules for the storage of biological reference standards. Matejtschuk P, Rafiq S, Johnes S, Gaines Das R. Biologicals. 2005 Jun;33(2):63-70. https://www.ncbi.nlm.nih.gov/pubmed/15939283
A big thank you to my co-authors of the soon-to-be released BEBPA White Paper on “Reference Standards for Potency Assays.” The discussions with this amazing group greatly influenced my thoughts presented in the article above. Any flawed logic or errors are all mine, any brilliance is theirs. These authors are: Jane Robinson, Dorota Bulik, Marie Gottar-Guillier, Matt Borer, Mike Merges, Mike Sadick, Peter Rigsby, Sian Estdale, and Seth Foltz.
The BEBPA white paper is currently entering the final stages of writing and will be available on the BEBPA website soon. A free webinar on the topic is happening November 15, 2023. Please join us by registering HERE.