
BEBPA Blog
Volume 1, Issue 1
Technical and Regulatory Considerations for Cell and Gene Therapy Raw Materials
By Laureen Little, PhD, President of BEBPA and Principal Consultant, Quality Services
Ref: 1
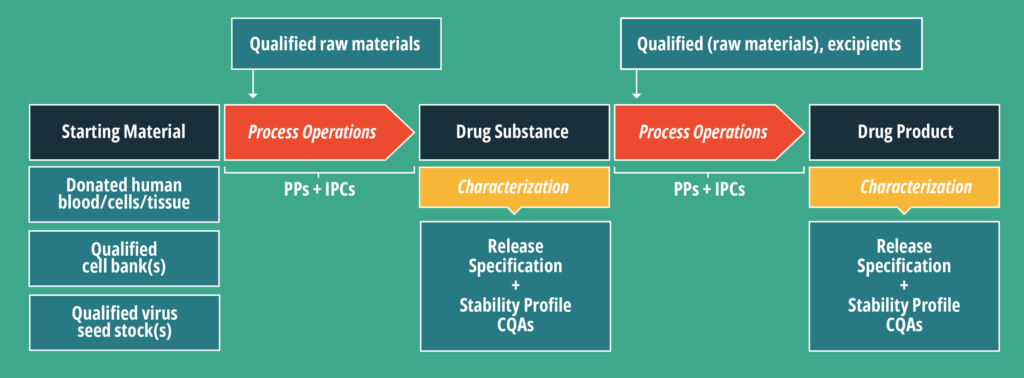
In Figure 1, three steps have material inputs. These can be delineated into two broad categories: Starting material (SM) and qualified raw materials, referred to in this article as ancillary materials (AM). There are several excellent references detailing appropriate risk assessments for various AMs, including those sourced from human and animal origins. (Ref: 2-7)
Starting material is more similar to an Active Pharmaceutical Ingredient (API) than an AM and is therefore more complex to source. The SM is expected to be in the final CGT product, therefore the regulations, like those of an API, are more detailed and specific.
CTGs have a variety of potential SMs depending on the product. As indicated in Figure 1 these might include:
- Donated human blood
- Donated human cells
- Donated human tissue
- Qualified cell banks
- Qualified virus seed stock(s)
The cell bank and virus seed stocks are SMs we routinely utilize in other classes of biotherapeutic products. As such, they have been thoroughly discussed in several recent publications. (Ref. 8 and 9)
However, less discussion can be found in the literature about the quality attributes and regulatory oversight required for SM sourced from donated human materials, specifically for CGT products. These donated materials are typically sourced from establishments that manufacture human cells, tissues, and cellular and tissue-based products (HCT/Ps) to supply medical facilities requiring material (e.g. blood, retinas, etc.) to treat patients as part of a medical procedure. These HCT/P facilities should comply with regulatory requirements as described in Title 21 Code of Federal Regulations, Part 1271 (21 CFR Part 1271), Subparts A-F. (Figure 2)

All HCT/P facilities, including collection facilities, must comply with subparts A through C, however, the need to comply with subpart D depends on the type of product being manufactured at that facility.
HCT/P facilities can be thought of manufacturing broadly two types of products:
- Those which are minimally manipulated and intended for homologous use (Note: Homologous is not the same as autologous. Homologous as defined by the FDA means “the repair, reconstruction, replacement, or supplementation of a recipient’s cells or tissues with an HCT/P that performs the same basic function or functions in the recipient as in the donor, including when such cells or tissues are for autologous use. This, therefore, covers blood donated by a donor for self-use, as well as blood donated for use by others.)
- Products which are manipulated to be products with a specified therapeutic mechanism of action.
The FDA has established two primary regulatory pathways for these two types of products. Under the first scheme, cellular therapy products that meet all the criteria in 21 CFR 1271.10(a) are regulated solely as HCT/Ps and are not required to be licensed, approved, or cleared. These products are commonly referred to as “361 products” because they are regulated solely under Section 361 of the Public Health Service (PHS) Act. The regulatory focus on these products is prevention of communicable disease. These facilities do not need to comply with Subpart D above.
If a cellular therapy product does not meet all the criteria above, it is regulated as a drug, device, and/or biological product under the Federal Food, Drug, and Cosmetic Act (FDCA) and Section 351 of the PHS Act, and as such, is commonly referred to as a “351 product.” (Figure 3)
As outlined in Figure 1, a CGT is a therapeutic product, which is manufactured according to the 21 CFR parts 200, 820 or 600 depending on whether the final product is considered to be a drug, biologic or device. However, the CGT has an SM which is considered a 361 HCT/P. This means that when thinking about sourcing SM for your CGT, it is important to remember that the HCT/P supplier must abide by the 1271 regulations.

If the donor SM provider is a collection facility which only provides 361 products, they must be registered and listed with the FDA as described in subparts A and B. Additionally, they need to follow Subpart C to establish donor eligibility. This includes the medical history of the donor as well as testing for various adventitious agents. However, there is no requirement for these facilities to follow the subpart D Good Tissue Practices. Also be aware that as a 361 facility, there are no biennial inspection requirements. This lack of mandated biennial inspections does not mean facilities are never inspected. It simply means it is not guaranteed that they are inspected biennially. According to the FDA compliance dashboard (https://datadashboard.fda.gov/ora/cd/inspections.htm), there were 422 inspections of HCT/P facilities in 2023.
A look at the for 438 observations issued by the FDA in 2023 to 361 product facilities reveal ongoing problems with assessing donor eligibility.
This clearly demonstrates that the starting/raw material sourcing group for a CGT needs to be aware that a simple statement that an HCT/P facility is registered and listed with the FDA does not assure compliance with donor eligibility requirements as required in Subpart C. I strongly recommended that raw material groups include subject matter experts in the technical requirements of the donated SM, as well as medical experts to assess potential clinical eligibility requirements for donors and compliance experts who understand the appropriate regulations for HCT/Ps.
Failure to recognize the strategic importance and complexities of carefully managing the SM and its suppliers can result in delays, costs, and failures in your CGT program.
References
- Manufacturing process development of ATMPs within a regulatory framework for EU clinical trial & marketing authorisation applications, Giulia Detela & Anthony Lodge, Cell Gene Therapy Insights 2016; 2(4), 425-452. https://www.insights.bio/immuno-oncology-insights/journal/article/467/manufacturing-process-development-of-atmps-within-a-regulatory-framework-for-eu-clinical-trial-marketing-authorisation-applications
- Ph.Eur. 5.2.12. Raw Materials of Biological Origin for the Production of Cell-Based and Gene Therapy Medicinal Products
- Ph.Eur. 5.1.7. Viral Safety
- USP <1043> Ancillary materials for cell, gene, and tissue-engineered products
- EudraLex: The Rules Governing Medicinal Products in the European Union Volume 4. Good Manufacturing Practice. Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products
- USP <1046> Cell-Based Advanced Therapies and Tissue-Based Products
- PDA Technical Report No. 81 (TR 81) Cell-Based Therapy Control Strategy. ISBN Number: 9781945584060)
- Project A-Gene, https://alliancerm.org/wp-content/uploads/2021/09/ALL-PROJECT-A-GENE-V10.pdf)
- Best Practices For Raw Material And Supplier Management For Cell & Gene Therapy Manufacturing, Andreas M. Beckhaus and David J. Cady, ProDeMaCon LLC Consulting, Cell & Gene, Guest Column | May 23, 2023, https://www.cellandgene.com/doc/best-practices-for-raw-material-and-supplier-management-for-cell-gene-therapy-manufacturing-0001)

About The Author: Laureen Little, Ph.D.
Dr. Laureen Little has over 30 years of biotechnology experience. She is the president of Biopharmaceutical Emerging Best Practices Association (BEBPA; www.bebpa.org) a non-profit organization she founded in in 2007 to promote scientific conferences for scientists working in the biopharmaceutical industry. BEBPA now hosts 3 to 4 conferences annually on such topics as potency bioassays, Host Cell Proteins, Immunogenicity assays, automation in analytical laboratories and the handling of reference materials. The organization has grown over the past decade and is now recognized as the venue for frank, open discussion and has resulted in vast improvements in technical approaches in various aspects of analytical development.
She is also a principal consultant with Quality Services, specializing in biological assay optimization and validation. She has been an instructor for FasTrain Courses (www.traincourses.com), for more than 20 years teaching various GXP courses as well as a popular potency assay course.
Laureen has worked with many firms developing, qualifying and validating biological potency assays for BLA submissions, Pre-Approval Supplements for commercial products and special amendments to support facility/manufacturing changes. The various products she has worked with include: monoclonal antibodies, rDNA products, peptide hormones, CBER regulated devices, gene therapy products, autologous cell therapies, toxins, cancer vaccines, viral vaccines, prophylactic vaccines, blood products, enzyme therapeutics, anti-angiogenic drugs, lipid-based therapies. The potency assays have included animal model systems, cell-based methods and binding assays.
BEBPA Categories
Email Updates
Welcome To BEBPA
Welcome to the BioPharmaceutical Emerging Best Practices Association (BEBPA), a non-profit organization managed by and for the benefit of the biopharmaceutical scientific community. Our focus is to provide a platform for industrial scientists to discuss common technical problems, suggest potential solutions and openly discuss the merits of such solutions.
BEBPA Conferences
2025 EUR BIOASSAY CONFERENCE
24-26 September 2025 ● Rotterdam, Netherlands ● Hybrid Conference
2026 US BIOASSAY CONFERENCE
March 23-25, 2025 ● College Park, Maryland ● Hybrid Conference
2026 HCP CONFERENCE
May 27-29, 2025 ● Boulder, Colorado ● Hybrid Conference
Stay Connected
OFFICE ADDRESS
P.O. Box 4458 Bremerton, WA 98312
OFFICE PHONE
206-651-4542
EMAIL ADDRESS
contactus@bebpa.org